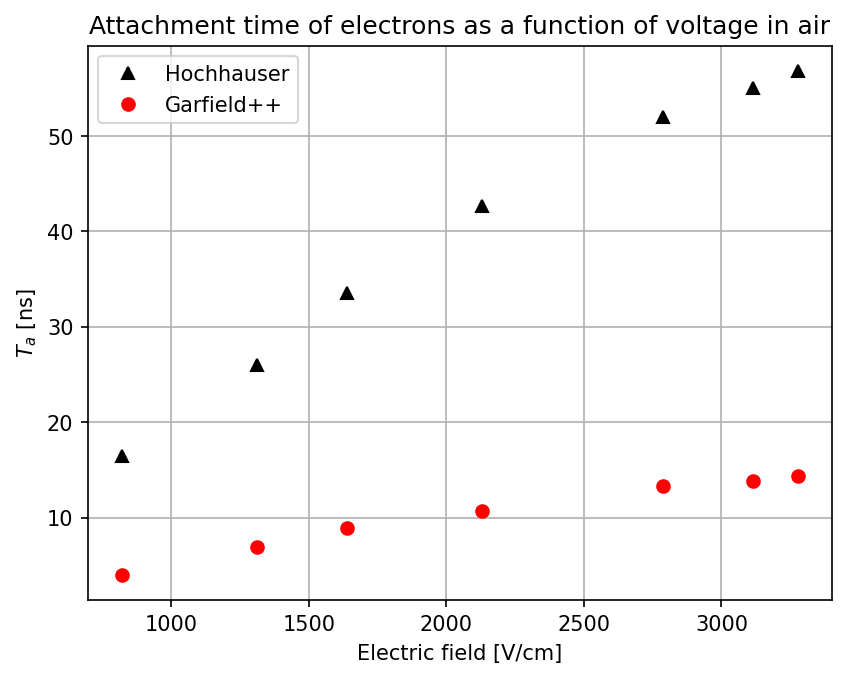
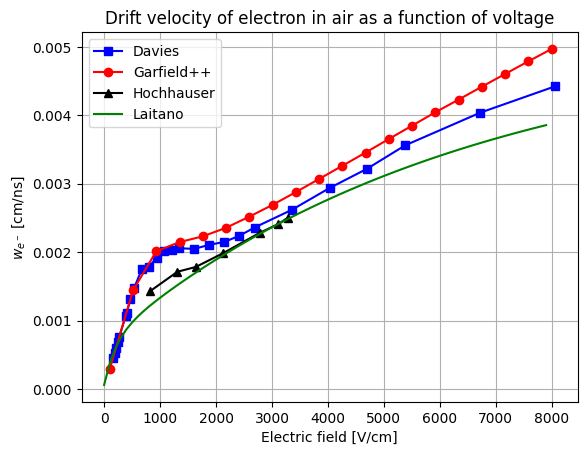
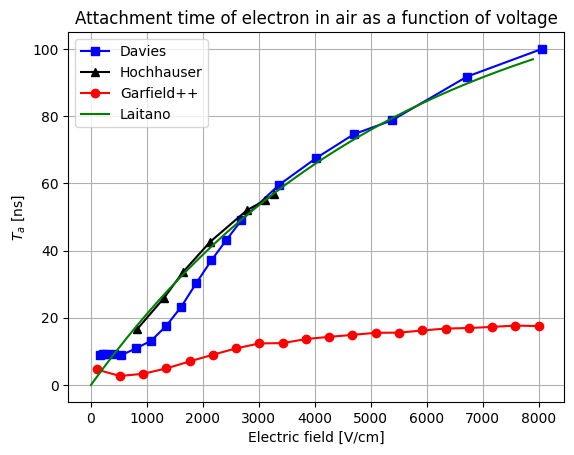
Dear experts,
I’m trying to validate what Garfield output by comparing to some experimental values by 2 ways.
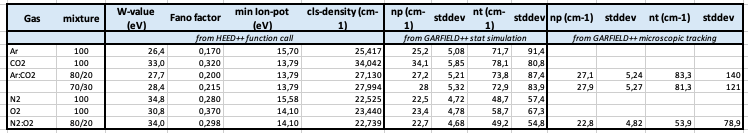
- First one, I’m trying to get the correct number of total electrons from a ionizing particle (proton) at MIP (\beta\gamma = 3.3) using TrackHeed in N2/O2 (80/20) mixture (with a gas file I have generated) and in Ar/CO2 (80/20) (with the gas file in the examples) both at NTP. Comparing to tables found online and in lectures, the values I get are quite far.
| Gas 1 | n_p (cm^{-1}) | n_t (cm^{-1}) |
|---|---|---|
| O2 | 22 | 73 |
| N2 | 10 | 56 |
| Ar | 23 | 94 |
| CO2 | 35.5 | 91 |
| — | — | — |
| N2/O2 (80/20) | 12.4 | 59.4 |
| Ar/CO2 (80/20) | 25.5 | 93.4 |
On 1000 tracks of proton in a plane parallel geometry with a gap of 0.1cm I get for total electrons in:
- N2/O2 (80/20): 7.09 so 70.9 cm^{-1}
- Ar/CO2 (80/20): 6.10 so 61.0 cm^{-1}
- O2: 6.99 so 69.9 cm^{-1}
Is it normal to have such a big difference from the literature ? (I mean O2 is ok but the 2 mixtures are not so accurate).
Here is my full code
const double gap = .1; // [cm]
const double detectorXWidth = 10; // [cm]
const double detectorZWidth = 10; // [cm]
const double vPlate = 500.; // [V]
string particle = "proton";
const double KineticEnergy = 230e6; // [eV] (230Mev or 230.e6 eV for proton therapy [eV])
const double BetaGamma = 3.3;
const unsigned int nTracks = 1000;
//vector<string> gasFile = {"ar_80_co2_20_0T.gas", "IonMobility_Ar+_Ar.txt"};
//vector<string> gasFile = {"air_merged.gas", "IonMobility_N2+_N2.txt", "IonMobility_O2-_air.txt"};
vector<string> gasFile = {"pureOxygen_merged.gas", "IonMobility_O2-_O2.txt"};"IonMobility_O2-_air.txt"};
//vector<string> gasFile = {"Hochhauser_N2_O2_100kPa.gas", "IonMobility_N2+_N2.txt", "IonMobility_O2-_air.txt"};
// Gas
MediumMagboltz gas;
gas.LoadGasFile(gasFile[0]);
const string path = getenv("GARFIELD_INSTALL");
gas.LoadIonMobility(path + "/share/Garfield/Data/" + gasFile[1]);
gas.LoadNegativeIonMobility(path + "/share/Garfield/Data/" + gasFile[2]);
// Geometry
ComponentAnalyticField cmp;
cmp.SetMedium(&gas);
cmp.AddPlaneY( 0., 0.);
cmp.AddPlaneY(gap, vPlate, "s");
// Sensor
Sensor sensor;
sensor.SetArea(-detectorXWidth/2, 0, -detectorZWidth/2, detectorXWidth/2, gap, detectorZWidth/2);
sensor.AddComponent(&cmp);
sensor.AddElectrode(&cmp, "s");
const double tstep = 1;
const double tmin = -0.5 * tstep;
const unsigned int nbins = 1000;
sensor.SetTimeWindow(tmin, tstep, nbins);
// Track
TrackHeed track;
track.SetSensor(&sensor);
track.SetParticle(particle);
//track.SetKineticEnergy(KineticEnergy);
track.SetBetaGamma(BetaGamma);
//track.Initialise(&gas, true);
// Avalanche for the electrons
AvalancheMicroscopic aval;
aval.SetSensor(&sensor);
// Drift for the ions
AvalancheMC drift;
drift.SetSensor(&sensor);
vector<int> vec_cluster;
vector<int> vec_totalElectron;
vector<int> vec_multiplicationElectron;
vector<int> vec_leftedElectron;
vector<int> vec_attachedElectron;
vector<int> vec_collectedElectron;
int flag_electronStatus = 0; // flag for the electron status in case of wierd status
for (unsigned int j = 0; j < nTracks; ++j) {
cout << "Track " << j+1 << "/" << nTracks << endl;
//sensor.NewSignal();
track.NewTrack(0, 0, 0, 0, 0, 1, 0); // x0, y0, z0, t0, dx0, dy0, dz0
int ncluster = 0; // number of clusters
int totalElectron = 0; // primary + delta electrons
int multiplicationElectron = 0; // primary + delta electrons after multiplication
int leftedElectron = 0; // electrons who left the drift area
int attachedElectron = 0; // electrons attached to a molecule/atom
int collectedElectron = 0; // electrons collected by the electrode
for (const auto& cluster : track.GetClusters()) {
ncluster ++;
totalElectron += cluster.electrons.size();
for (const auto& electron : cluster.electrons) {
aval.AvalancheElectron(electron.x, electron.y, electron.z, electron.t, 0.1, 0, 0, 0); // x0, y0, z0, t0, e0, dx0, dy0, dz0
const unsigned int np = aval.GetNumberOfElectronEndpoints();
double xe1, ye1, ze1, te1, e1;
double xe2, ye2, ze2, te2, e2;
int status;
multiplicationElectron += np;
for (unsigned int j = 0; j < np; ++j) {
aval.GetElectronEndpoint(j, xe1, ye1, ze1, te1, e1, xe2, ye2, ze2, te2, e2, status);
//drift.DriftIon(xe1, ye1, ze1, te1);
if (status == -7) {
attachedElectron ++;
//drift.DriftNegativeIon(xe2, ye2, ze2, te2);
}
if (status == -5) collectedElectron ++;
if (status == -1) leftedElectron ++;
if (status == -3 || status == -8 || status == -16 || status == -17) flag_electronStatus +=1;
}
}
}
vec_cluster.push_back(ncluster);
vec_totalElectron.push_back(totalElectron);
vec_multiplicationElectron.push_back(multiplicationElectron);
vec_leftedElectron.push_back(leftedElectron);
vec_attachedElectron.push_back(attachedElectron);
vec_collectedElectron.push_back(collectedElectron);
}
double mean_cluster = accumulate(vec_cluster.begin(), vec_cluster.end(), 0.0) / nTracks;
double mean_totalElectron = accumulate(vec_totalElectron.begin(), vec_totalElectron.end(), 0.0) / nTracks;
int absolute_multiplicationElectron = accumulate(vec_multiplicationElectron.begin(), vec_multiplicationElectron.end(), 0.0) - accumulate(vec_totalElectron.begin(), vec_totalElectron.end(), 0.0);
double mean_multiplicationElectron = accumulate(vec_multiplicationElectron.begin(), vec_multiplicationElectron.end(), 0.0) / nTracks;
double mean_leftedElectron = accumulate(vec_leftedElectron.begin(), vec_leftedElectron.end(), 0.0) / nTracks;
double mean_attachedElectron = accumulate(vec_attachedElectron.begin(), vec_attachedElectron.end(), 0.0) / nTracks;
double mean_collectedElectron = accumulate(vec_collectedElectron.begin(), vec_collectedElectron.end(), 0.0) / nTracks;
auto stop = chrono::high_resolution_clock::now();
auto duration = chrono::duration_cast<chrono::seconds>(stop - start);
time_t now = time(0);
char* dt = ctime(&now);
cout << endl;
cout << " " << dt << endl;
cout << "Gap size: " << gap << " cm" << endl;
cout << "Voltage: " << vPlate << " V" << endl;
cout << "Particle: " << particle << endl;
cout << "Kinetic energy: " << KineticEnergy << " eV/c" << endl;
cout << "Gas file: " << gasFile[0] << endl;
cout << " " << gasFile[1] << endl;
cout << " " << gasFile[2] << endl;
cout << endl;
cout << " " << "Mean on " << nTracks << " different tracks" << endl;
cout << "Clusters: " << mean_cluster << endl;
cout << "Total electrons: " << mean_totalElectron << endl;
cout << "Total electrons after avalanche: " << mean_multiplicationElectron << endl;
cout << "Multiplication (absolute): " << "+" << absolute_multiplicationElectron << endl;
cout << "Ratio of attached electrons: " << double (mean_attachedElectron/mean_multiplicationElectron) << endl;
cout << "Ratio of collected electrons: " << double (mean_collectedElectron/mean_multiplicationElectron) << endl;
cout << "Ratio of electrons who left the drift area: " << double (mean_leftedElectron/mean_multiplicationElectron) << endl;
if (flag_electronStatus > 0) cout << "/!\\ There was a problem computing the electron status /!\\"<< endl;
cout << endl;
cout << "Time taken by computation: " << duration.count() << " seconds" << endl << endl;
I also tried to turn off the simulation of the \delta electrons (secondary electrons) with track.DisableDeltaElectronTransport() add simulate those secondary electrons with AvalancheMicroscopic and the primaries electorns in input.
for (const auto& cluster : track.GetClusters()) {
ncluster ++;
primaryElectron += cluster.electrons.size();
for (const auto& electron : cluster.electrons) {
aval.AvalancheElectron(electron.x, electron.y, electron.z, electron.t, electron.e, electron.dx, electron.dy, electron.dz); // x0, y0, z0, t0, e0, dx0, dy0, dz0
const unsigned int np = aval.GetNumberOfElectronEndpoints();
double xe1, ye1, ze1, te1, e1;
double xe2, ye2, ze2, te2, e2;
int status;
totalElectron += np;
for (unsigned int j = 0; j < np; ++j) {
aval.GetElectronEndpoint(j, xe1, ye1, ze1, te1, e1, xe2, ye2, ze2, te2, e2, status);
//drift.DriftIon(xe1, ye1, ze1, te1);
if (status == -7) {
attachedElectron ++;
//drift.DriftNegativeIon(xe2, ye2, ze2, te2);
}
if (status == -5) collectedElectron ++;
if (status == -1) leftedElectron ++;
if (status == -3 || status == -8 || status == -16 || status == -17) flag_electronStatus +=1;
}
}
}
With this method the computation takes longer and I have significant different results. Which method is more correct ?
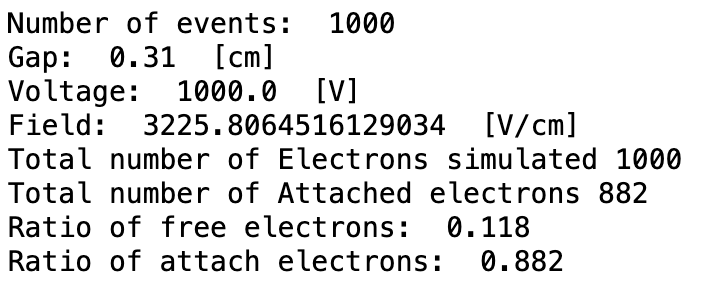
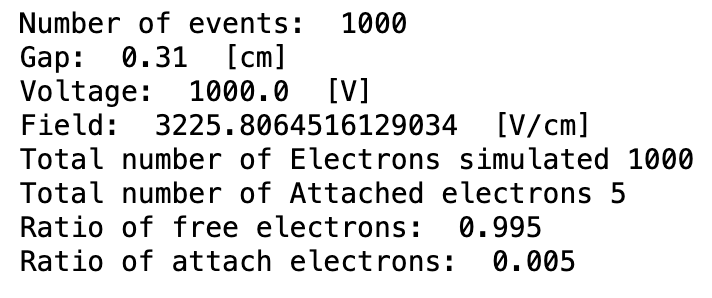
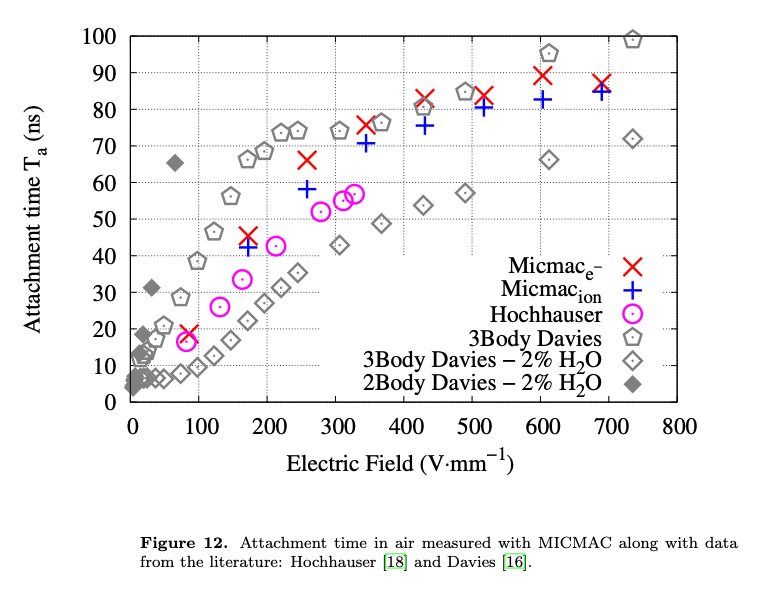
- The second way is by comparing the number of collected electrons with the free electron fraction (i.e. the fraction of electron who escape the recombination and reach the electrode as free electron) measured by Hochhauser that show J.W. Boag in is paper of 1996 ( DOI 10.1088/0031-9155/41/5/005). In his paper we have 2 plots for gaps of 0.31 cm and 0.61 cm both in dry syntethic air and laboratory air.
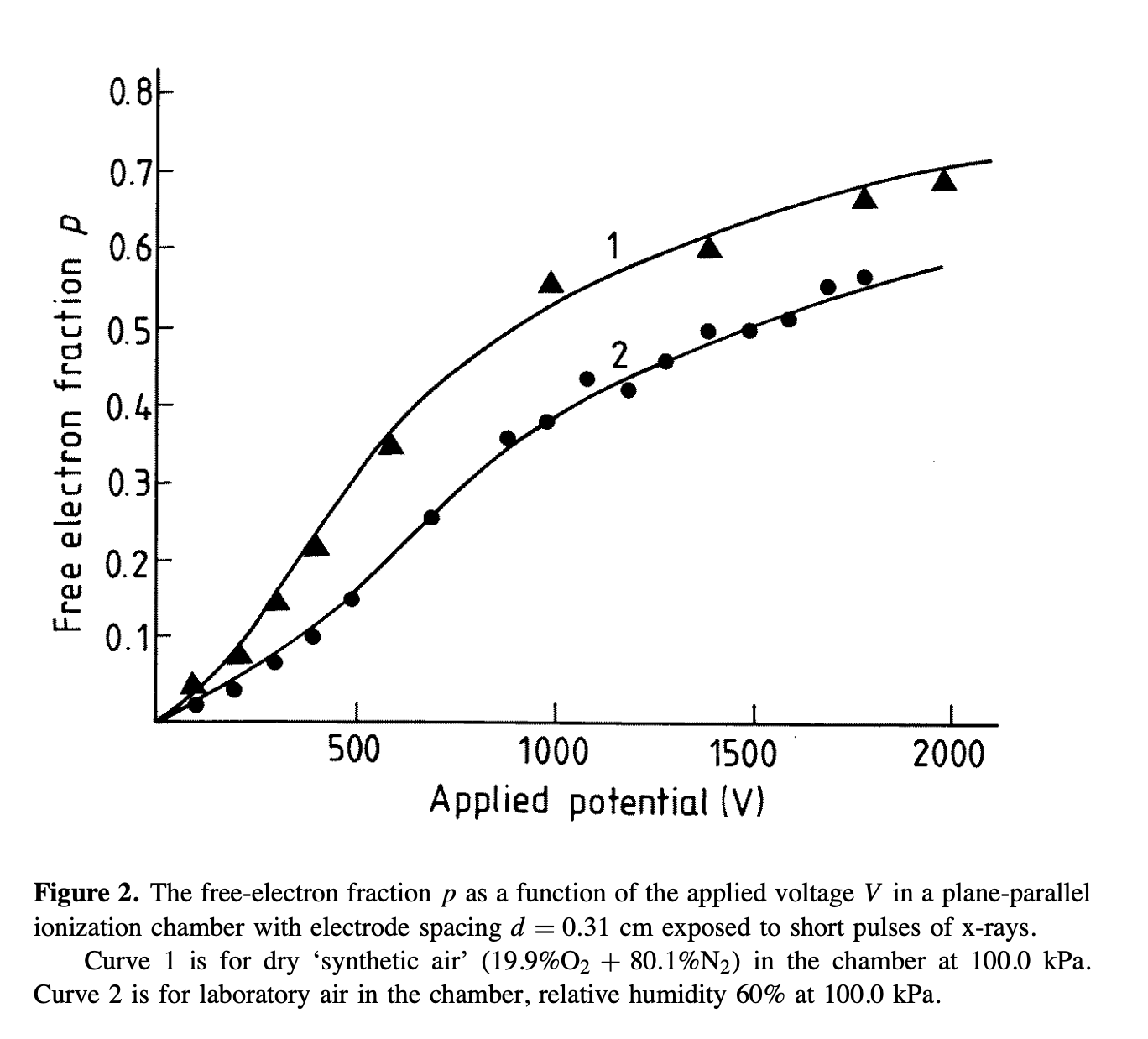
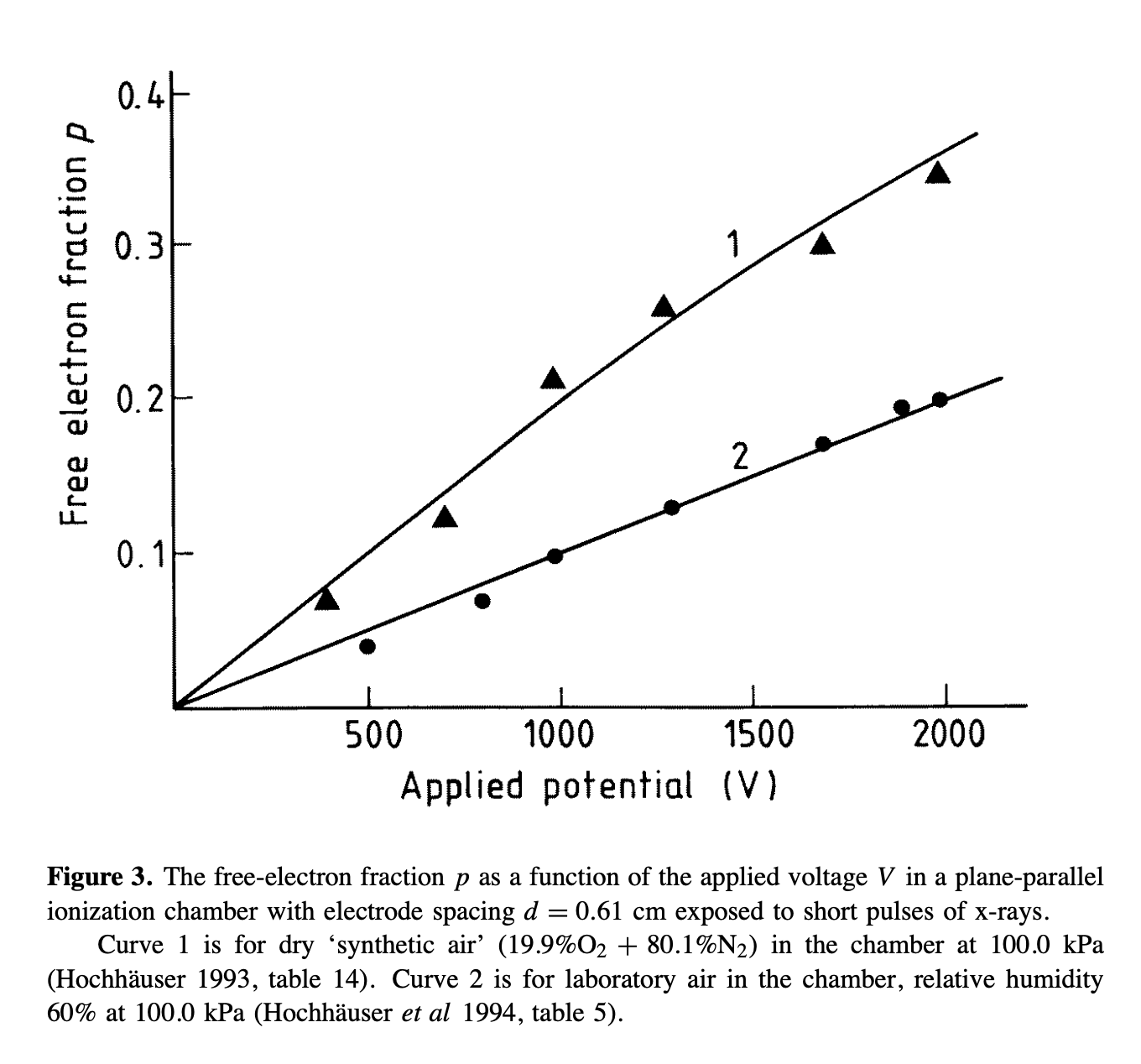
I have generate a new gas file matching the synthetic air at 100 kPa to compare. With this code, and for both gaps i tried get the value of the collected electrons by putting electrons uniformly distributed in the plane parallel geometry at different potential but as you seen the results from Garfield are not matching Hochhauser measurement.
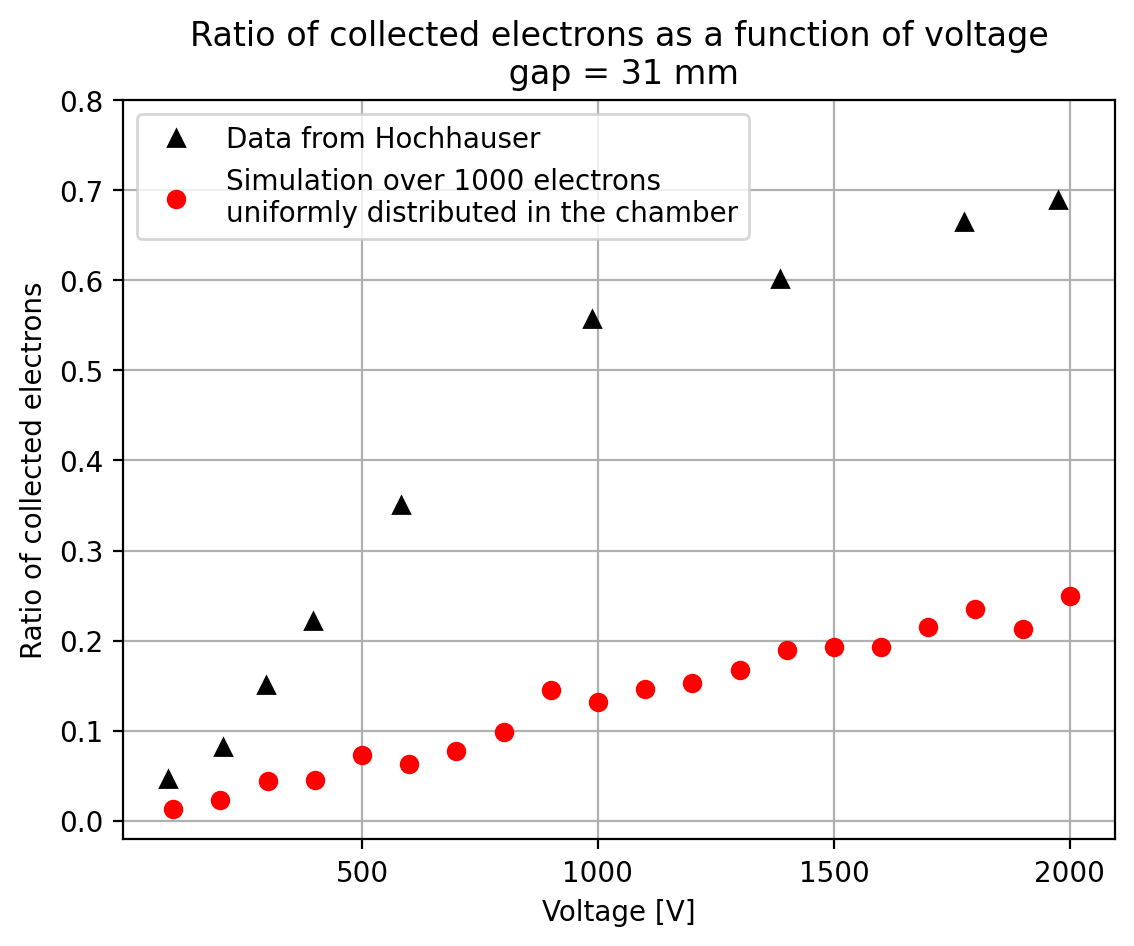
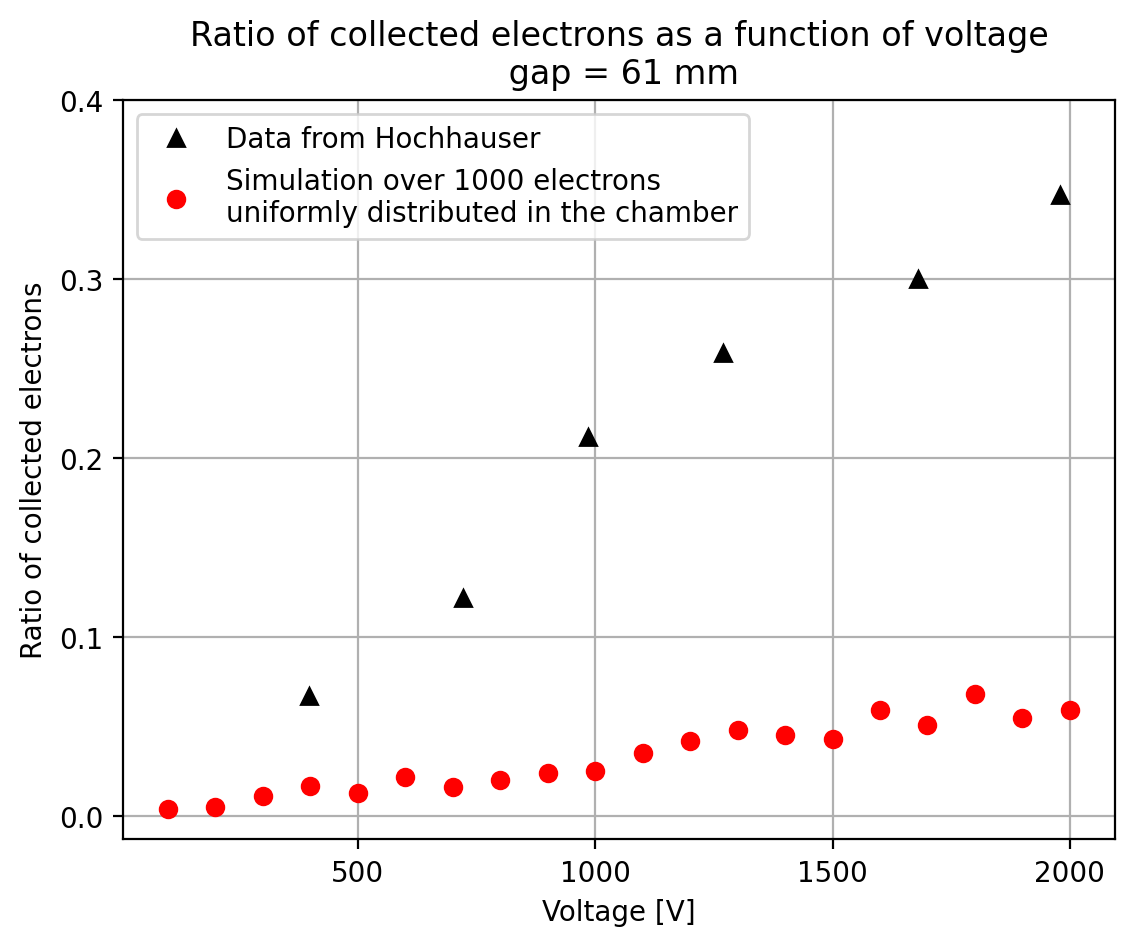
Thanks a lot for your help.
Pierre
vector<double> vPlateValues = {100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1100,
1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000};
for (double vPlate : vPlateValues) {
const double gap = .31; // [cm]
const double detectorXWidth = 5; // [cm]
const double detectorZWidth = 5; // [cm]
const unsigned int nElectrons = 1000;
vector<string> gasFile = {"Hochhauser_N2_O2_100kPa_NO_HUMID.gas",
"IonMobility_N2+_N2.txt",
"IonMobility_O2-_air.txt"};
// Gas
MediumMagboltz gas;
gas.LoadGasFile(gasFile[0]);
const string path = getenv("GARFIELD_INSTALL");
gas.LoadIonMobility(path + "/share/Garfield/Data/" + gasFile[1]);
gas.LoadNegativeIonMobility(path + "/share/Garfield/Data/" + gasFile[2]);
// Geometry
ComponentAnalyticField cmp;
cmp.SetMedium(&gas);
cmp.AddPlaneY( 0., 0.);
cmp.AddPlaneY(gap, vPlate, "s");
// Sensor
Sensor sensor;
sensor.SetArea(-detectorXWidth/2, 0, -detectorZWidth/2, detectorXWidth/2, gap, detectorZWidth/2);
sensor.AddComponent(&cmp);
sensor.AddElectrode(&cmp, "s");
const double tstep = 1;
const double tmin = -0.5 * tstep;
const unsigned int nbins = 1000;
sensor.SetTimeWindow(tmin, tstep, nbins);
// Avalanche for the electrons
AvalancheMicroscopic aval;
aval.SetSensor(&sensor);
// Drift for the ions
AvalancheMC drift;
drift.SetSensor(&sensor);
vector<int> vec_totalElectron;
vector<int> vec_leftedElectron;
vector<int> vec_attachedElectron;
vector<int> vec_collectedElectron;
int flag_electronStatus = 0; // flag for the electron status in case of weird status
for (unsigned int j = 0; j < nElectrons; ++j) {
cout << "Electron " << j+1 << "/" << nElectrons << endl;
int totalElectron = 0; // primary + delta electrons + avalanche if any
int leftedElectron = 0; // electrons who left the drift area
int attachedElectron = 0; // electrons attached to a ion
int collectedElectron = 0; // electrons collected by the electrode
double x0 = -detectorXWidth/2 + detectorXWidth * RndmUniform();
double y0 = gap * RndmUniform();
double z0 = -detectorZWidth/2 + detectorZWidth * RndmUniform();
aval.AvalancheElectron(x0, y0, z0, 0, 0.1, 0, 0, 0); // x0, y0, z0, t0, e0, dx0, dy0, dz0
const unsigned int np = aval.GetNumberOfElectronEndpoints();
double xe1, ye1, ze1, te1, e1;
double xe2, ye2, ze2, te2, e2;
int status;
totalElectron += np;
for (unsigned int j = 0; j < np; ++j) {
aval.GetElectronEndpoint(j, xe1, ye1, ze1, te1, e1, xe2, ye2, ze2, te2, e2, status);
//drift.DriftIon(xe1, ye1, ze1, te1);
if (status == -7) {
attachedElectron ++;
//drift.DriftNegativeIon(xe2, ye2, ze2, te2);
}
if (status == -5) collectedElectron ++;
if (status == -1) leftedElectron ++;
if (status == -3 || status == -8 || status == -16 || status == -17) flag_electronStatus +=1;
}
vec_totalElectron.push_back(totalElectron);
vec_leftedElectron.push_back(leftedElectron);
vec_attachedElectron.push_back(attachedElectron);
vec_collectedElectron.push_back(collectedElectron);
}
double mean_totalElectron = accumulate(vec_totalElectron.begin(), vec_totalElectron.end(), 0.0) / nElectrons;
double mean_leftedElectron = accumulate(vec_leftedElectron.begin(), vec_leftedElectron.end(), 0.0) / nElectrons;
double mean_attachedElectron = accumulate(vec_attachedElectron.begin(), vec_attachedElectron.end(), 0.0) / nElectrons;
double mean_collectedElectron = accumulate(vec_collectedElectron.begin(), vec_collectedElectron.end(), 0.0) / nElectrons;
// Time
auto stop = chrono::high_resolution_clock::now();
auto duration = chrono::duration_cast<chrono::seconds>(stop - start);
time_t now = time(0);
char* dt = ctime(&now);
cout << endl;
cout << " " << dt << endl;
cout << "Gap size: " << gap << " cm" << endl;
cout << "Voltage: " << vPlate << " V" << endl;
cout << "Gas file: " << gasFile[0] << endl;
cout << " " << gasFile[1] << endl;
cout << " " << gasFile[2] << endl;
cout << endl;
cout << " " << "Mean on " << nElectrons << " different tracks" << endl;
cout << "Total electrons: " << mean_totalElectron << endl;
cout << "Ratio of attached electrons: " << double (mean_attachedElectron/mean_totalElectron) << endl;
cout << "Ratio of collected electrons: " << double (mean_collectedElectron/mean_totalElectron) << endl;
cout << "Ratio of electrons who left the drift area: " << double (mean_leftedElectron/mean_totalElectron) << endl;
if (flag_electronStatus > 0) cout << "/!\\ There was a problem computing the electron status /!\\"<< endl;
cout << endl;
cout << "Time taken by computation: " << duration.count() << " seconds" << endl << endl;