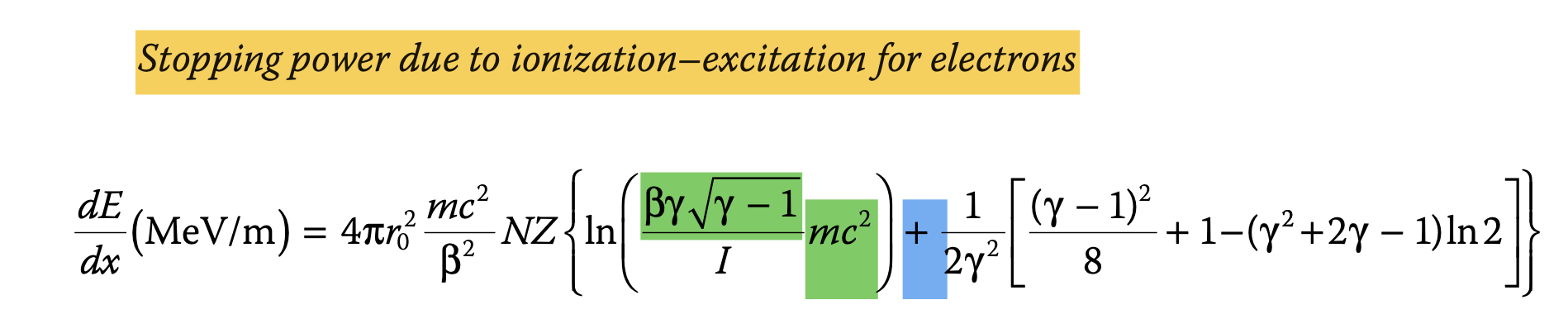Hi all,
I couldn’t see the mistake . Please help me , why the code cannot do the fitting.
Cheers.
codes:
Si_estar.txt (6.4 KB)
fittingcode3.cpp (4.8 KB)
source.h (1.6 KB)
Hi all,
I couldn’t see the mistake . Please help me , why the code cannot do the fitting.
Cheers.
codes:
Si_estar.txt (6.4 KB)
fittingcode3.cpp (4.8 KB)
source.h (1.6 KB)
What you sent gives:
root [0]
Processing fittingcode3.cpp...
In file included from input_line_8:1:
/Users/couet/Downloads/fittingcode3.cpp:30:10: fatal error: '.../Header.h' file not found
#include ".../Header.h"
^~~~~~~~~~~~~~
root [1]
I changed the original one, let me recheck.
Did you put your path for the header file!
#include “…/source.h”. inside the second file.
I ran your example without any changes . Can you provide an example ready to run ?
fittingcode3.cpp (4.8 KB)
source.h (1.6 KB)
In fittingcode3.cpp file, you have to put your path. Updated file above!
Still an other file is missing:
root [0]
Processing fittingcode3.cpp...
Error in <TGraph::TGraph>: Cannot open file: /Users/spyhunter0066/Library/CloudStorage/OneDrive-harran.edu.tr/MacbookCplusplus/RangeCalculators/Fitting/Si_estar.txt, TGraph is Zombie
I tried to play a bit with your macro by changing the initial value of the parameter and also adding one parameter (an offset, as it seems your fit is to low on Y) but that didi not help. May be you function cannot fit these data ? lets ping @moneta . He may have more idea.
I simplified you macro to:
const double PI = 3.141592653589793;
const double Avogadro = 6.022E23; //atoms/mole
const double c_square = 931.5 ; //MeV/u
const double FourPiRsquare= 4.0* PI * (2.818*1.E-15) * (2.818*1.E-15); //m^2
const double m_electron = 0.00054858 ; //in akb, u; 9.11E-31; //kg
double A_Si = 28.0 ;
double rho_Si = 2.33*1.E3 ;
double N_Si = (rho_Si*Avogadro)/ (A_Si*1.0E-3);
double Z_Si = 14.0 ;
double MEP_Si = 172.*1.0E-6 ;
TF1* f_T_particle;
TF1* f_LorentzFactor_e ;
TF1* f_Beta_e;
TF1* f_STP_e;
TF1* f_STP_e_over_rho;
double MEP_medium = MEP_Si; // MEP_Calculator(Z_Si); //mean potential energy
double RME_calculator(double m_particle);
double T_particle(double *x,double *par);
double LorentzFactor_e(double *x,double *par);
double Beta_e(double *x, double *par);
double STP_e(double *x, double *par);
double STP_e_over_rho(double *x, double *par);
double RME_e = RME_calculator(m_electron); // rest mass energy
void fittingcode3() {
auto c1 = new TCanvas("c1", "c1",294,78,1343,770);
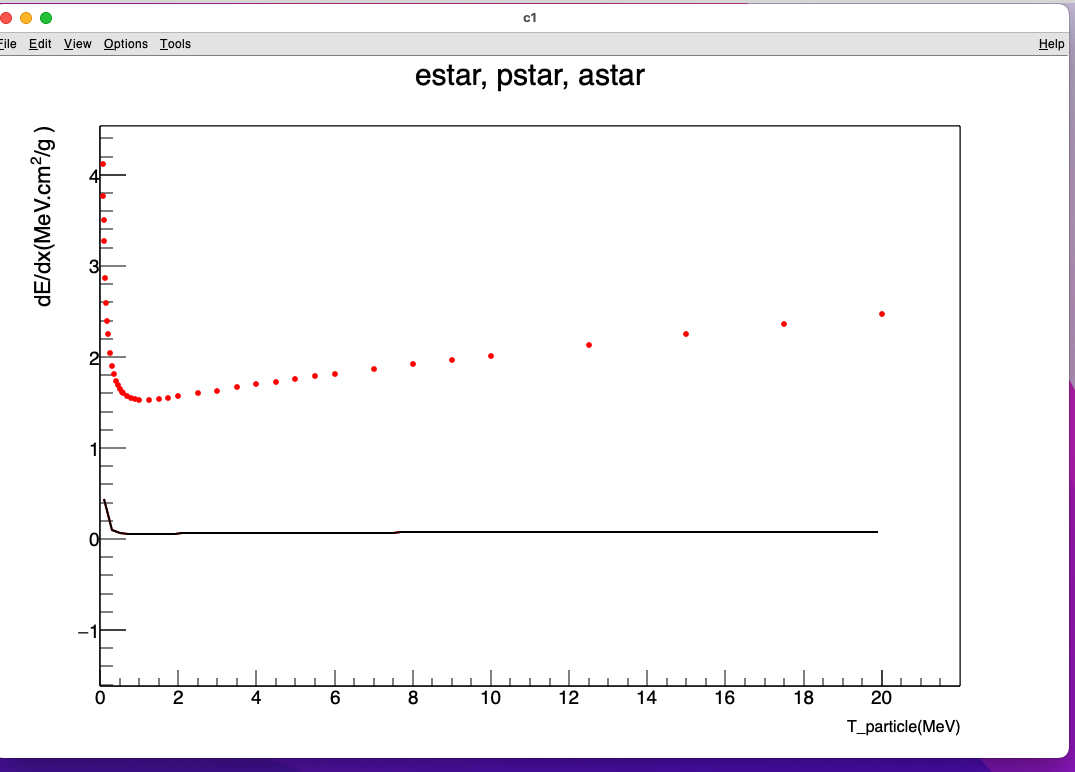
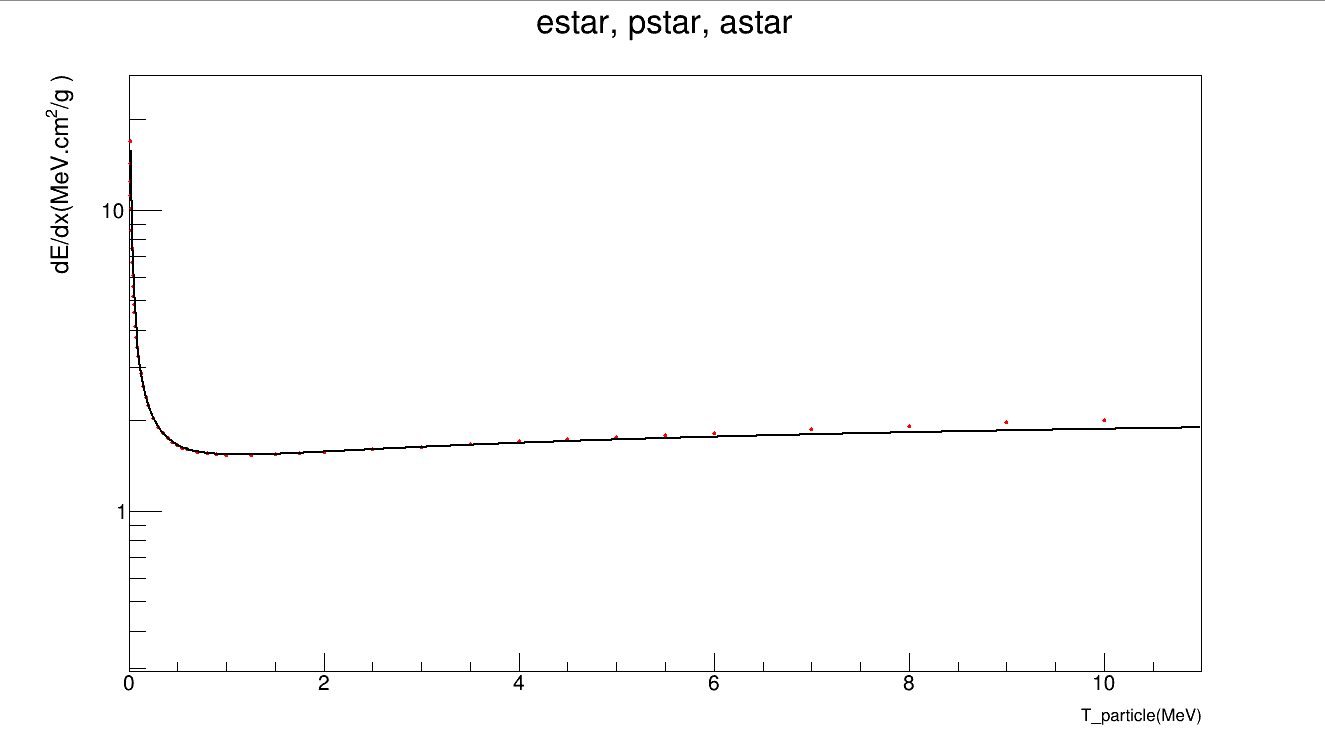
auto gr1 = new TGraph("Si_estar.txt"); //total STP (MeV. cm^2 /g)
gr1->SetMarkerSize(0.6);
gr1->SetMarkerStyle(20);
gr1->SetMarkerColor(kRed);
gr1->Draw("AP");
gr1->GetXaxis()->SetRangeUser(1.e-2, 20.);
gr1->GetYaxis()->SetRangeUser(1.e-2, 50.);
double min_Limit= 1.e-2;
double max_Limit= 20. ;
f_T_particle = new TF1("f_T_particle", T_particle, min_Limit,max_Limit,0);
f_LorentzFactor_e = new TF1("f_LorentzFactor_e", LorentzFactor_e, min_Limit,max_Limit,0 ); //between 1 and 10
f_Beta_e = new TF1("f_Beta_e",Beta_e, min_Limit,max_Limit,0); //between 0 and 1
f_STP_e = new TF1("f_STP_e", STP_e, min_Limit, max_Limit,0);
f_STP_e_over_rho= new TF1("f_STP_e_over_rho", STP_e_over_rho, min_Limit, max_Limit,1);
f_STP_e_over_rho->SetParameter(0, 0.);
// f_STP_e_over_rho->SetParameter(1, 0.);
gr1->Fit(f_STP_e_over_rho, "MR");
}
double T_particle(double *x,double *par){
return x[0];
}
double RME_calculator(double m_particle){
double RME;
RME= m_particle * c_square; //u* (MeV/u) = MeV
return RME; //MeV
}
double LorentzFactor_e(double *x,double *par){
double result1= ((x[0] / RME_e) +1.0);
return result1;
}
double Beta_e(double *x, double *par){
double result2= sqrt( (pow(pow(x[0]/RME_e,2.) +1. ,2.) -1. ) /(pow(pow(x[0]/RME_e,2.) +1. ,2.)) ) ;
return result2;
}
double STP_e(double *x,double *par){ //Unit is MeV/m
double result3 = FourPiRsquare* RME_e* N_Si * Z_Si*pow(1./f_Beta_e->Eval(x[0]),2)* (log(f_Beta_e->Eval(x[0])*f_LorentzFactor_e->Eval(x[0])*sqrt(f_LorentzFactor_e->Eval(x[0])-1.) /MEP_medium) +((1.0/(2.0*pow(f_LorentzFactor_e->Eval(x[0]),2.)))*((pow(f_LorentzFactor_e->Eval(x[0])-1., 2.)/8.0)+1.0-(log(2.)*(pow(f_LorentzFactor_e->Eval(x[0]),2.)+(2.*f_LorentzFactor_e->Eval(x[0]))-1.)))));
return result3 ;
}
double STP_e_over_rho(double *x,double *par){ // (MeV .m^2/kg) * 10 = (MeV.cm^2 /g)
double arg = 10. * (f_STP_e->Eval(x[0]) / rho_Si);
printf("par[0] =%f\n",par[0]);
return par[0] * arg ; //MeV * cm^2 /g
}
@moneta
I suspected the initial parameter value as well. However, I couldn’t succeed. Function shape looks right. Database values on TGraph seems high. Functions are from the book.
I kept the parameter changing a bit. Interestingly, the fit didn’t move an inch! Why!
f_STP_e_over_rho->SetParameter(0, 30.);
So I spent a bit of time to understand where the error was.
The error was in the function were you define the Beta of the electron from its kinetic energy
What you have is
double result2= sqrt( (pow(pow(x[0]/RME_e,2.) +1. ,2.) -1. ) /(pow(pow(x[0]/RME_e,2.) +1. ,2.)) ) ;
what you want instead is
double result2= sqrt( (pow(x[0]/RME_e+1. ,2.) -1. ) /(pow(x[0]/RME_e +1. ,2.)) ) ;
Moreover in your final formula inside the logarithm you missed the electron rest mass, the change in the fit parameter is quite relevant it pass from 0.878 to 0.9987.
Below the result after of the fit after solving the issue with the beta factor
PS:
The MEP_medium of silicon that I found in the PDG is 173 eV and not 172 ,the effect of this parameter on the fit is very small.
I suggest you to rewrite the function for the stopping power in this way, you have less function and the readability of your code, according to me, is quite better.
double STP_e(double *x,double *par){ //Unit is MeV/m
double T=x[0];
double gamma=(T/RME_e) +1.;
double beta = sqrt(1.-1./gamma/gamma);
double result3 = FourPiRsquare* RME_e* N_Si * Z_Si * 1. /pow(beta,2) * ( log( beta*gamma*sqrt(gamma-1.)*RME_e/MEP_medium) + 1./2./pow(gamma,2) * ( pow(gamma-1,2.)/8. +1. -log(2.)*(pow(gamma,2) +2. *gamma -1.)));
return result3 ;
}
Thanks. Since the formula is long, apparently I missed some of it.
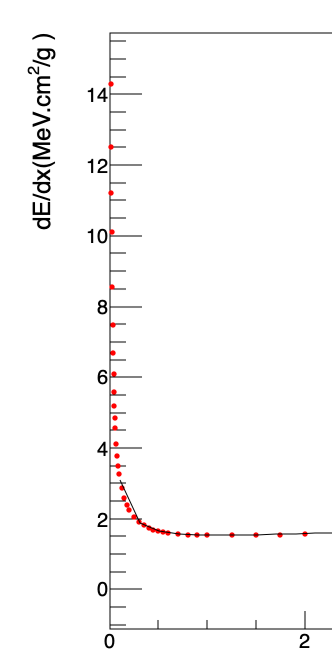
@Dilicus : When I zoomed in, I noticed the function didn’t start from 0.01 MeV in my code. Why is that? Yours looks fine.
I changed a bit the code, I set the number of points of the function to be 500 and I set the x minimum to 5e-3.
For instance, why it does not start from the the number I set up ? I wanted graph, and all functions to have the same range on X axis; however, it doesn’t seem like they do by looking at the line. We shouldn’t need to set it up like you did. They should all start from 1.e-2 as I told it so.
Another issue is when I try to zoom in the plot in TBrowser. It does not allow me to zoom in , for instance, between 0 and 2 on X axis. Why? I does not even let me do right click on my mouse to set the X range.
These are the code, I used I changed a bit few parameters like the MEP of silicon, the electron mass multiplied by c square is already in MeV.
I restored the limit of your function.
I do not know why you cannot zoom in TBrowser.
fittingcode3.cpp (4.6 KB)
source.h (1.6 KB)
Thanks all.
I realized " f_STP_e_over_rho->SetNpx(50000.); " is an option to widen the points in the fit for a function. Otherwise, it does not do it automatically.
I still have no idea why I cannot focus on TBrowser with my mouse click on X axis. Maybe a glitch.
@Dilicus
Is there any automated way to get the X axis range where the fit is perfect instead of me trying to change and find the range THE FIT is possible? Most of the time, I waste time when finding the initial values of Xrange where the fit works. It also misleads me most of the time by letting me think the code is wrong instead of the range I set initially is not suitable.
For instance, it cannot do the fit if I lower the X min limit close to zero, so I have to start somewhere close to 1.
Cheers.
I didn’t had the time to look in your code carefully yesteraday. But I think you were using a Bethe-Bloch. The range of application of the Bethe Bloch goes from \beta\gamma\sim0.05 to \beta\gamma\sim80. Given that your X axis is on kinetic energy, you should set the fit range according to the particle mass.
Not the original Bethe Bloch, but something similar.
I think I should check where the function is defined and restricted by the logarithmic term.
dE/dx term should be positive at all times.
Formula References:
double T_particle(double *x,double *par){
return x[0];
}
double min_Limit= 0. ;
double max_Limit= 1. ;
TF1* f_T_particle = new TF1(“f_T_particle”, T_particle, min_Limit,max_Limit,0);
f_T_particle->SetNpx(1000.);
int Numberx = f_T_particle->GetNpx();
Is there any way to create a loop to print out all the energies incremented 0.001 precision?
double T[Numberx];
for(int i=0; i < Numberx; i++){
T[i] = f_T_particle->Eval(i);
}
Normally, the f_T_particle function should have infinite amount of real numbers on X axis, but we can set it up by using “SetNpx()” function. When I try to see what numbers on my X range, it gets blurry. I can only get integer numbers due to the limitation in for loop incrementation.
Can I see all the numbers in 0.001 increment between 0 and 1 for instance?
Change just the for loop to
for(double i=0; i < 1; i+=0.001){
T[i] = f_T_particle->Eval(i);
}
or
for(int i=0; i < Numberx; i++){
T[i] = f_T_particle->Eval(double(i)/dobule(Numberx));
}