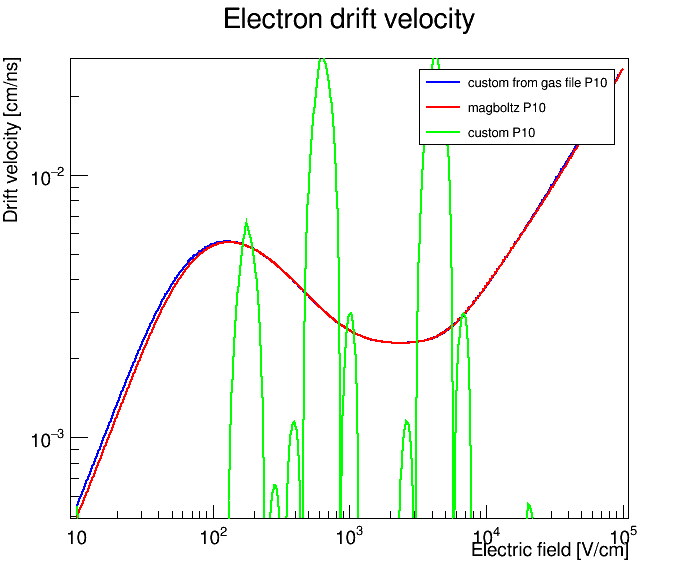
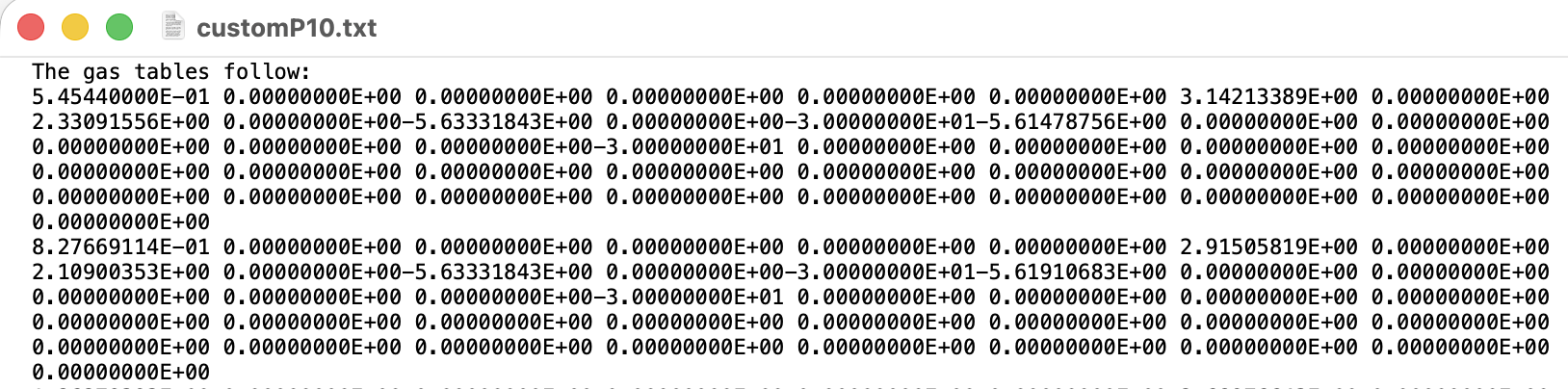
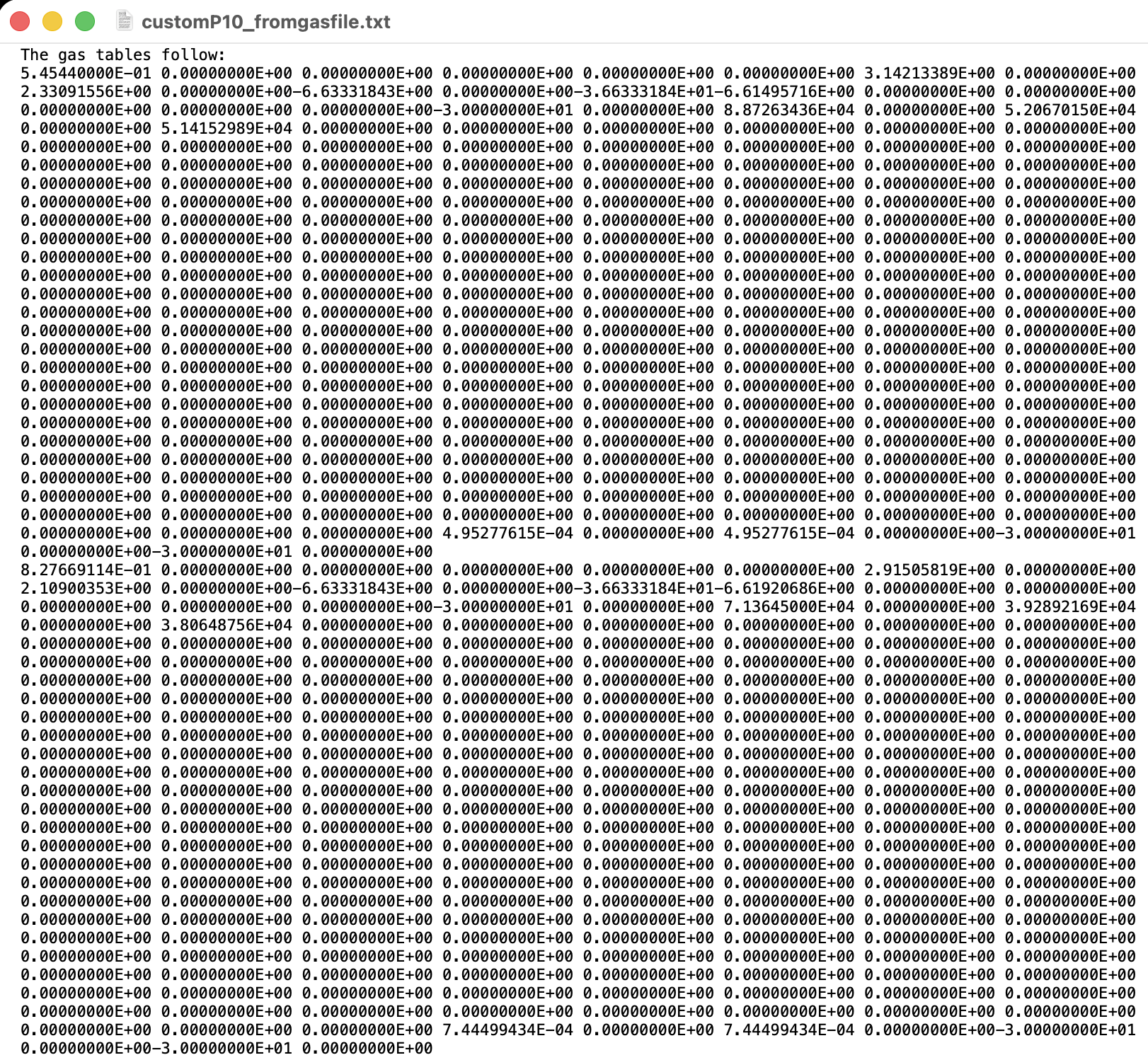
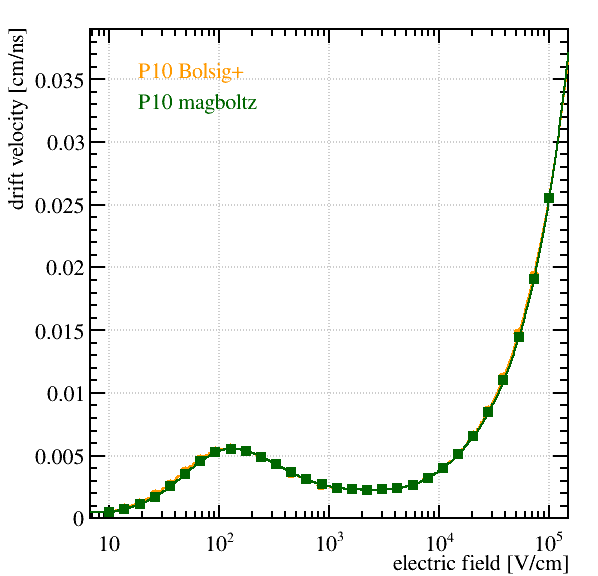
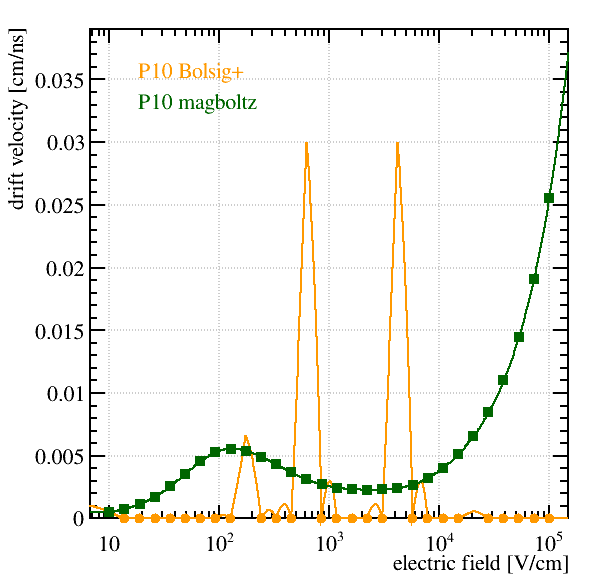
Hi all,
thanks a lot for the helpful feedback so far — I really appreciate the time and suggestions.
Unfortunately, I am a new user on the forum and I am not yet able to upload attachments(I think). I will add the files as soon as that becomes possible, but in the meantime I can show the code of the converter and the .gas file.
The Garfield-specific part of the converter starts at around line 330, where I construct and fill the MediumGas object with the BOLSIG+ transport parameters.The lines before this part of the code are only used to parse and handle the BOLSIG+ output files.
#include <algorithm>
#include <cctype>
#include <cmath>
#include <cstdlib>
#include <fstream>
#include <iomanip>
#include <iostream>
#include <map>
#include <regex>
#include <set>
#include <sstream>
#include <stdexcept>
#include <string>
#include <vector>
#include "Garfield/MediumGas.hh"
namespace {
constexpr double kBoltzmann = 1.380649e-23; // J/K
constexpr double kTorrToPa = 133.32236842105263; // Pa / Torr
constexpr double kPi = 3.14159265358979323846;
// ---------- utilities ----------
std::string Trim(const std::string& s) {
size_t i = 0, j = s.size();
while (i < j && std::isspace(static_cast<unsigned char>(s[i]))) ++i;
while (j > i && std::isspace(static_cast<unsigned char>(s[j - 1]))) --j;
return s.substr(i, j - i);
}
std::string Lower(std::string s) {
for (char& c : s) c = static_cast<char>(std::tolower(static_cast<unsigned char>(c)));
return s;
}
std::string NormalizeLabel(std::string s) {
s = Lower(Trim(s));
s = std::regex_replace(s, std::regex(R"(\s+)"), " ");
return s;
}
struct TransportRow {
double en_td;
double muN;
double alpha_over_N;
double eta_over_N;
double dL_N;
double dT_N;
};
bool ParseDoubleToken(const std::string& token, double& out) {
char* end = nullptr;
out = std::strtod(token.c_str(), &end);
return end != token.c_str() && *end == '\0';
}
std::vector<double> ExtractNumbers(const std::string& line) {
std::vector<double> values;
std::stringstream ss(line);
std::string tok;
while (ss >> tok) {
double x = 0.;
if (ParseDoubleToken(tok, x)) values.push_back(x);
}
return values;
}
bool IsMostlyNumericLine(const std::string& line) {
auto nums = ExtractNumbers(line);
if (nums.empty()) return false;
// Require at least one numeric token and no alphabetic chars.
for (char c : line) {
if (std::isalpha(static_cast<unsigned char>(c))) return false;
}
return true;
}
double GasNumberDensity(double pressure_torr, double temperature_K) {
const double p_pa = pressure_torr * kTorrToPa;
return p_pa / (kBoltzmann * temperature_K); // m^-3
}
// BOLSIG+ reduced field: E/N in Td, with 1 Td = 1e-21 V m^2.
// Garfield++ grid needs E in V/cm.
double ENTdToEfieldVcm(double en_td, double N) {
return en_td * 1.e-23 * N;
}
// BOLSIG+ mobility*N is in 1/(m V s).
// v = mu * E = (muN / N) * (E/N * N) = muN * (E/N).
double DriftVelocityCmNs(double muN, double en_td) {
const double en_si = en_td * 1.e-21; // V m^2
const double v_m_s = muN * en_si; // m/s
return v_m_s * 1.e-7; // cm/ns
}
// alpha/N, eta/N in m^2 -> alpha, eta in cm^-1
double CoeffCmInv(double coeff_over_N, double N) {
return coeff_over_N * N * 1.e-2;
}
// D*N in 1/(m s) -> D in m^2/s
double ReducedDiffusionToSI(double DN, double N) {
return DN / N;
}
// Garfield++ wants diffusion coefficients in sqrt(cm). The user guide gives
// dl, dt in sqrt(cm). A consistent conversion from a conventional diffusion
// constant D uses sqrt(2D/v)
double GarfieldDiffusionSqrtCm(double D_m2_s, double v_cm_ns) {
if (v_cm_ns <= 0.) return 0.;
const double D_cm2_ns = D_m2_s * 1.e-5;
const double coeff_cm = 2.0 * D_cm2_ns / v_cm_ns;
return coeff_cm > 0. ? std::sqrt(coeff_cm) : 0.;
}
// ---------- block parser ----------
// files are block-style:
// Label line
// one or more numeric lines
// next label line
//
// parse into: label -> concatenated numeric array.
std::map<std::string, std::vector<double>> ParseParameterBlocks(const std::string& filename) {
std::ifstream fin(filename);
if (!fin) throw std::runtime_error("Cannot open file: " + filename);
std::map<std::string, std::vector<double>> blocks;
std::string line, currentLabel;
while (std::getline(fin, line)) {
line = Trim(line);
if (line.empty()) continue;
auto nums = ExtractNumbers(line);
std::stringstream ss(line);
std::string tok;
int nTokens = 0;
int nNumeric = 0;
while (ss >> tok) {
++nTokens;
double x = 0.;
if (ParseDoubleToken(tok, x)) ++nNumeric;
}
// If almost all tokens are numeric, treat as data.
if (nTokens > 0 && nNumeric == nTokens) {
if (!currentLabel.empty()) {
auto& dest = blocks[currentLabel];
dest.insert(dest.end(), nums.begin(), nums.end());
}
continue;
}
// Otherwise treat as a label.
currentLabel = NormalizeLabel(line);
if (!blocks.count(currentLabel)) {
blocks[currentLabel] = {};
}
}
return blocks;
}
const std::vector<double>& GetRequiredBlock(
const std::map<std::string, std::vector<double>>& blocks,
const std::vector<std::string>& candidateLabels,
const std::string& fileTag) {
for (const auto& label : candidateLabels) {
auto it = blocks.find(NormalizeLabel(label));
if (it != blocks.end() && !it->second.empty()) return it->second;
}
std::ostringstream os;
os << "Could not find required block in " << fileTag << ". Tried labels:";
for (const auto& s : candidateLabels) os << "\n - " << s;
os << "\n\nAvailable parsed labels in " << fileTag << ":";
for (const auto& kv : blocks) {
os << "\n * [" << kv.first << "] (" << kv.second.size() << " values)";
}
throw std::runtime_error(os.str());
}
void CheckSameSize(const std::vector<std::pair<std::string, size_t>>& sizes) {
if (sizes.empty()) return;
const size_t n = sizes.front().second;
for (const auto& x : sizes) {
if (x.second != n) {
std::ostringstream os;
os << "Array size mismatch:\n";
for (const auto& y : sizes) os << " " << y.first << ": " << y.second << "\n";
throw std::runtime_error(os.str());
}
}
}
void Usage(const char* argv0) {
std::cerr
<< "Usage:\n"
<< " " << argv0
<< " output_PT.dat output_SST.dat output_gn_PT.dat output.gas pressure_torr temperature_K\n\n"
<< "Example:\n"
<< " " << argv0
<< " output_PT.dat output_SST.dat output_gn_PT.dat custom.gas 760 293.15\n";
}
} // namespace
// to get all the transport parameters, I need three different runs with BOLSIG+ with different settings/growth models=> get three output files
// parse three outfiles. The E/N grid is the same in all the runs. I only take the E/N values from the output_PT file.
int main(int argc, char* argv[]) {
if (argc != 7) {
Usage(argv[0]);
return 1;
}
const std::string filePT = argv[1];
const std::string fileSST = argv[2];
const std::string fileGNPT = argv[3];
const std::string outGas = argv[4];
const double pressure_torr = std::stod(argv[5]);
const double temperature_K = std::stod(argv[6]);
try {
const auto pt = ParseParameterBlocks(filePT);
const auto sst = ParseParameterBlocks(fileSST);
const auto gnpt = ParseParameterBlocks(fileGNPT);
// PT file
const auto& en_td = GetRequiredBlock(
pt,
{
"Electric field / N (Td)"
},
"output_PT.dat");
const auto& muN = GetRequiredBlock(
pt,
{
"Mobility *N (1/m/V/s)"
},
"output_PT.dat");
// SST file
const auto& alpha_over_N = GetRequiredBlock(
sst,
{
"Townsend ioniz. coef. alpha/N (m2)"
},
"output_SST.dat");
const auto& eta_over_N = GetRequiredBlock(
sst,
{
"Townsend attach. coef. eta/N (m2)"
},
"output_SST.dat");
// gnPT file
const auto& dL_N = GetRequiredBlock(
gnpt,
{
"Longitud. diffusion coef. *N (1/m/s)"
},
"output_gnPT.dat");
const auto& dT_N = GetRequiredBlock(
gnpt,
{
"Diffusion coefficient *N (1/m/s)"
},
"output_gnPT.dat");
CheckSameSize({
{"E/N", en_td.size()},
{"Mobility*N", muN.size()},
{"alpha/N", alpha_over_N.size()},
{"eta/N", eta_over_N.size()},
{"DL*N", dL_N.size()},
{"DT*N", dT_N.size()}
});
const size_t n = en_td.size();
const double N = GasNumberDensity(pressure_torr, temperature_K);
std::vector<TransportRow> rows;
rows.reserve(n);
for (size_t i = 0; i < n; ++i) {
rows.push_back({
en_td[i],
muN[i],
alpha_over_N[i],
eta_over_N[i],
dL_N[i],
dT_N[i]
});
}
std::sort(rows.begin(), rows.end(),
[](const TransportRow& a, const TransportRow& b) {
return a.en_td < b.en_td;
});
std::vector<TransportRow> uniqueRows;
uniqueRows.reserve(rows.size());
for (const auto& r : rows) {
if (uniqueRows.empty() || r.en_td > uniqueRows.back().en_td) {
uniqueRows.push_back(r);
} else if (r.en_td == uniqueRows.back().en_td) {
std::cerr << "Warning: duplicate E/N value removed: "
<< r.en_td << " Td\n";
} else {
std::cerr << "Warning: non-monotonic E/N value skipped: "
<< r.en_td << " Td\n";
}
}
std::vector<double> efields;
efields.reserve(uniqueRows.size());
for (const auto& r : uniqueRows) {
efields.push_back(ENTdToEfieldVcm(r.en_td, N));
}
//Garfield related stuff happens here.
Garfield::MediumGas gas;
gas.PrintGas();
gas.SetPressure(pressure_torr);
gas.SetTemperature(temperature_K);
for (size_t i = 1; i < efields.size(); ++i) {
if (efields[i] <= efields[i - 1]) {
std::cerr << "Non-ascending E-field at i=" << i
<< ": " << efields[i - 1] << " then " << efields[i] << "\n";
}
}
gas.SetFieldGrid(efields, std::vector<double>{0.0}, std::vector<double>{0.5 * kPi});
for (size_t i = 0; i < uniqueRows.size(); ++i) {
const auto& r = uniqueRows[i];
const double vE = DriftVelocityCmNs(r.muN, r.en_td);
const double vB = 0.0;
const double vExB = 0.0;
const double DL_SI = ReducedDiffusionToSI(r.dL_N, N);
const double DT_SI = ReducedDiffusionToSI(r.dT_N, N);
const double dl = GarfieldDiffusionSqrtCm(DL_SI, vE);
const double dt = GarfieldDiffusionSqrtCm(DT_SI, vE);
const double alpha = CoeffCmInv(r.alpha_over_N, N);
const double eta = CoeffCmInv(r.eta_over_N, N);
const size_t ib = 0;
const size_t ia = 0;
std::cout << "E index: " << i << "\n";
std::cout << "E field: " << efields[i] << "\n";
std::cout << "drift: " << vE << "\n";
std::cout << "dl: " << dl << "\n";
std::cout << "dt: " << dt << "\n";
std::cout << "alpha: " << alpha << "\n";
std::cout << "eta: " << eta << "\n";
// set velocity
if (!gas.SetElectronVelocityE(i, ib, ia, vE)) {
throw std::runtime_error("SetElectronVelocityE failed at row " + std::to_string(i));
}
// check if the set velocity
double v = 0;
gas.GetElectronVelocityE(i, ib, ia, v);
std::cout << "test drift: " << v << "\n";
if (!gas.SetElectronVelocityB(i, ib, ia, vB)) {
throw std::runtime_error("SetElectronVelocityB failed at row " + std::to_string(i));
}
if (!gas.SetElectronVelocityExB(i, ib, ia, vExB)) {
throw std::runtime_error("SetElectronVelocityExB failed at row " + std::to_string(i));
}
if (!gas.SetElectronLongitudinalDiffusion(i, ib, ia, dl)) {
throw std::runtime_error("SetElectronLongitudinalDiffusion failed at row " + std::to_string(i));
}
if (!gas.SetElectronTransverseDiffusion(i, ib, ia, dt)) {
throw std::runtime_error("SetElectronTransverseDiffusion failed at row " + std::to_string(i));
}
if (!gas.SetElectronTownsend(i, ib, ia, alpha)) {
throw std::runtime_error("SetElectronTownsend failed at row " + std::to_string(i));
}
if (!gas.SetElectronAttachment(i, ib, ia,eta)) {
throw std::runtime_error("SetElectronAttachment failed at row " + std::to_string(i));
}
}
gas.PrintGas();
gas.WriteGasFile(outGas);
double vcheck;
std::cout << "test " << gas.GetElectronVelocityE(100., 0., 0., vcheck)<<"\nS";
std::cout << "Wrote " << outGas << "\n";
std::cout << "Rows: " << n << "\n";
std::cout << "Pressure [Torr]: " << pressure_torr << "\n";
std::cout << "Temperature [K]: " << temperature_K << "\n";
std::cout << "Number density [m^-3]: " << std::setprecision(12) << N << "\n";
std::cout << "E/N range [Td]: " << en_td.front() << " -> " << en_td.back() << "\n";
std::cout << "E range [V/cm]: " << efields.front() << " -> " << efields.back() << "\n";
} catch (const std::exception& e) {
std::cerr << "Error: " << e.what() << "\n";
return 2;
}
return 0;
}
And the first lines of the resulting .gas look like this.
*----.----1----.----2----.----3----.----4----.----5----.----6----.----7----.----8----.----9----.---10----.---11----.---12----.---13--
% Created 13/04/26 at 13.42.14 < none > GAS "none "
Version : 13
GASOK bits: TFTTFTFTTTFFFFFFFFFF
Identifier: Ar 100%, T=293.15 K, p=1 atm
Clusters :
Dimension : F 249 1 1 0 0
E fields
3.21883328E-03 3.41054699E-03 3.61368756E-03 3.82889875E-03 4.05695309E-03
4.29859090E-03 4.55461690E-03 4.82590017E-03 5.11334198E-03 5.41787580E-03
5.74056383E-03 6.08246831E-03 6.44474799E-03 6.82859386E-03 7.23532563E-03
7.66626303E-03 8.12285453E-03 8.60664517E-03 9.11927656E-03 9.66242249E-03
1.02379177E-02 1.08476613E-02 1.14937777E-02 1.21783270E-02 1.29036910E-02
1.36722197E-02 1.44865523E-02 1.53493606E-02 1.62635736E-02 1.72322493E-02
1.82586065E-02 1.93460893E-02 2.04983350E-02 2.17192063E-02 2.30128232E-02
2.43834668E-02 2.58357400E-02 2.73745354E-02 2.90049711E-02 3.07325189E-02
3.25630050E-02 3.45023520E-02 3.65572552E-02 3.87347959E-02 4.10417337E-02
4.34861157E-02 4.60763109E-02 4.88206881E-02 5.17282602E-02 5.48093274E-02
5.80738681E-02 6.15325045E-02 6.51974681E-02 6.90806685E-02 7.31953031E-02
7.75545690E-02 8.21739167E-02 8.70681527E-02 9.22540150E-02 9.77485634E-02
1.03570467E-01 1.09739039E-01 1.16275202E-01 1.23200522E-01 1.30538496E-01
1.38313266E-01 1.46551548E-01 1.55280058E-01 1.64528410E-01 1.74327826E-01
1.84710816E-01 1.95712145E-01 2.07368828E-01 2.19719813E-01 2.32806301E-01
2.46672391E-01 2.61364434E-01 2.76931034E-01 2.93425301E-01 3.10901634E-01
3.29418617E-01 3.49040624E-01 3.69827850E-01 3.91854326E-01 4.15194086E-01
4.39924382E-01 4.66125685E-01 4.93888122E-01 5.23305039E-01 5.54473002E-01
5.87495012E-01 6.22486949E-01 6.59561471E-01 6.98847331E-01 7.40470064E-01
7.84571299E-01 8.31302320E-01 8.80814414E-01 9.33274958E-01 9.88860990E-01
1.04775920E+00 1.11016272E+00 1.17628400E+00 1.24634512E+00 1.32057785E+00
1.39923005E+00 1.48256886E+00 1.57087111E+00 1.66443294E+00 1.76356657E+00
1.86860675E+00 1.97990113E+00 2.09782309E+00 2.22276854E+00 2.35515593E+00
2.49543269E+00 2.64406231E+00 2.80154051E+00 2.96840161E+00 3.14519925E+00
3.33252247E+00 3.53102792E+00 3.74131430E+00 3.96415412E+00 4.20025555E+00
4.45042327E+00 4.71549419E+00 4.99633739E+00 5.29395072E+00 5.60923544E+00
5.94331814E+00 6.29732543E+00 6.67238388E+00 7.06981322E+00 7.49086881E+00
7.93703129E+00 8.40978133E+00 8.91066398E+00 9.44138521E+00 1.00037154E+01
1.05995214E+01 1.12308312E+01 1.18997369E+01 1.26084918E+01 1.33594456E+01
1.41551412E+01 1.49982180E+01 1.58915408E+01 1.68380388E+01 1.78408985E+01
1.89035319E+01 2.00294154E+01 2.12223794E+01 2.24863830E+01 2.38256752E+01
2.52447300E+01 2.67483114E+01 2.83414408E+01 3.00294935E+01 3.18180382E+01
3.37130941E+01 3.57210023E+01 3.78486511E+01 4.01028001E+01 4.24914962E+01
4.50221430E+01 4.77037530E+01 5.05450171E+01 5.35555919E+01 5.67451338E+01
6.01249087E+01 6.37061826E+01 6.75005433E+01 7.15208660E+01 7.57806700E+01
8.02941180E+01 8.50763386E+01 9.01434260E+01 9.55124399E+01 1.01201406E+02
1.07228671E+02 1.13615480E+02 1.20382433E+02 1.27552384E+02 1.35149475E+02
1.43198811E+02 1.51727754E+02 1.60764950E+02 1.70340013E+02 1.80485454E+02
1.91235070E+02 2.02625233E+02 2.14693605E+02 2.27480742E+02 2.41029455E+02
2.55385451E+02 2.70596048E+02 2.86713068E+02 3.03789622E+02 3.21883328E+02
3.37369135E+02 3.53601711E+02 3.70616464E+02 3.88445581E+02 4.07137346E+02
4.26723947E+02 4.47256884E+02 4.68774785E+02 4.91329149E+02 5.14968261E+02
5.39743621E+02 5.65713168E+02 5.92931622E+02 6.21460141E+02 6.51359883E+02
6.82698444E+02 7.15546638E+02 7.49972060E+02 7.86055181E+02 8.23876472E+02
8.63516404E+02 9.05061885E+02 9.48606261E+02 9.94246098E+02 1.04208440E+03
1.09222095E+03 1.14477162E+03 1.19984907E+03 1.25757885E+03 1.31808648E+03
1.38150071E+03 1.44796962E+03 1.51763805E+03 1.59065406E+03 1.66718504E+03
1.74740158E+03 1.83147429E+03 1.91958985E+03 2.01194784E+03 2.10875103E+03
2.21020865E+03 2.31654925E+03 2.42800457E+03 2.54482247E+03 2.66726366E+03
2.79559211E+03 2.93009750E+03 3.07107274E+03 3.21883328E+03
E-B angles
1.57079633E+00
B fields
0.00000000E+00
...
a few things in the generated .gas file seem a bit odd:
- The gas is identified as Argon, although I never explicitly specified or set Argon anywhere in the conversion process.
- The electric field values listed in the
.gas file do not match the values I obtain when constructing the field grid in my program.
I am not sure if these observations are relevant, but they seemed a bit strange and might indicate that I am misunderstanding how the .gas format or the MediumGas interface is supposed to be used.
Thanks again for your help!
Best regards
Max